Case Studies
Covid-19 peptide inhibitors delivered in 6 months
2020.1
2020.2
2020.3
2020.4
2020.6
2020.7
Kick Off
Drug design
Candidate library design and ultra fast synthesis of 29 compounds
S2 protein binding & pseudo virus experiment: 12 active compounds
Test on live virus in P3 lab : 9 active compounds
Confirm 3 PCC & Client delivery
IND
Challenges
- As the global pandemic evolve rapidly, finding investigational treatments for Covid-19 is a race with time
- Data on the new virus is sparse and incomplete
Workflow
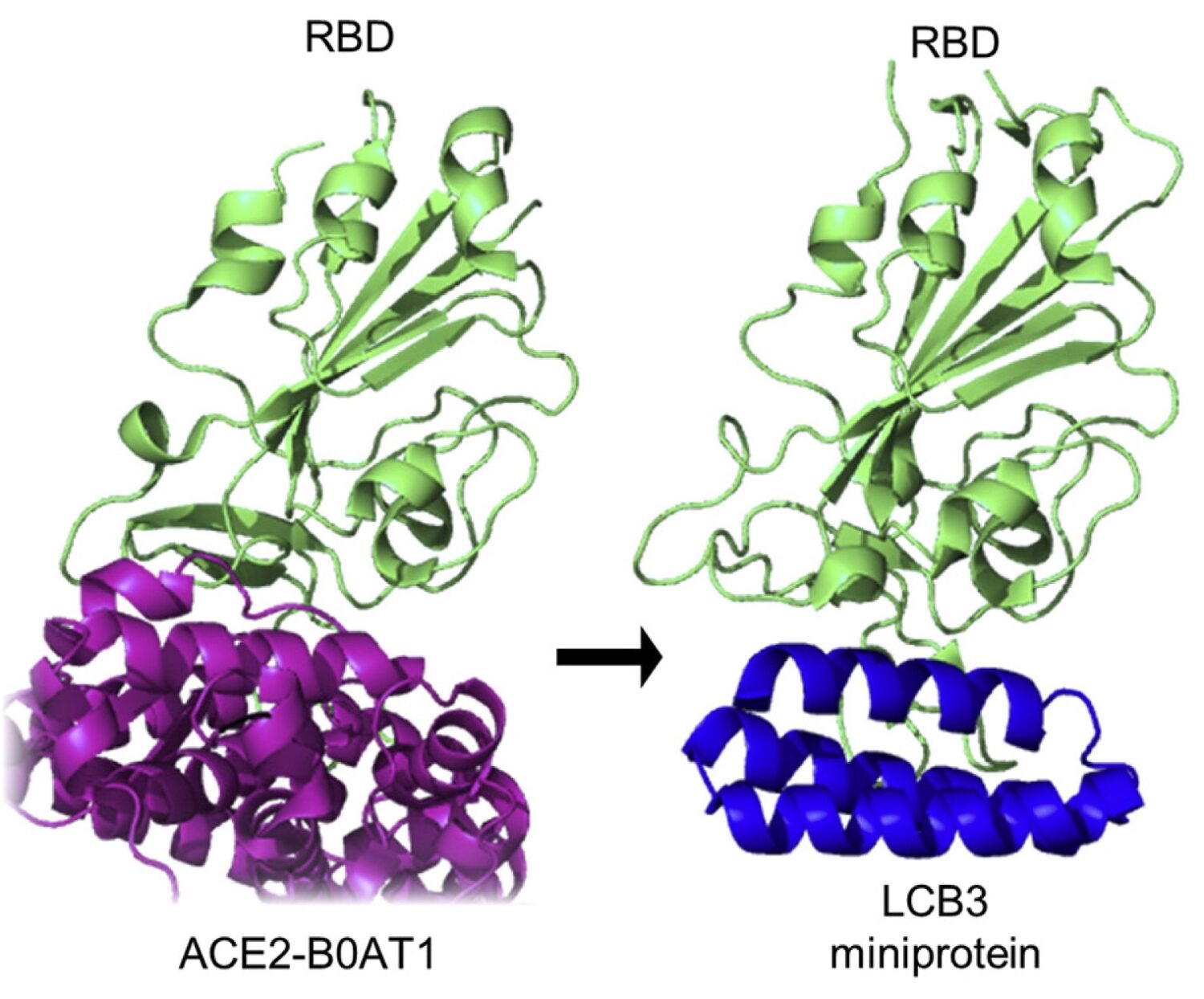
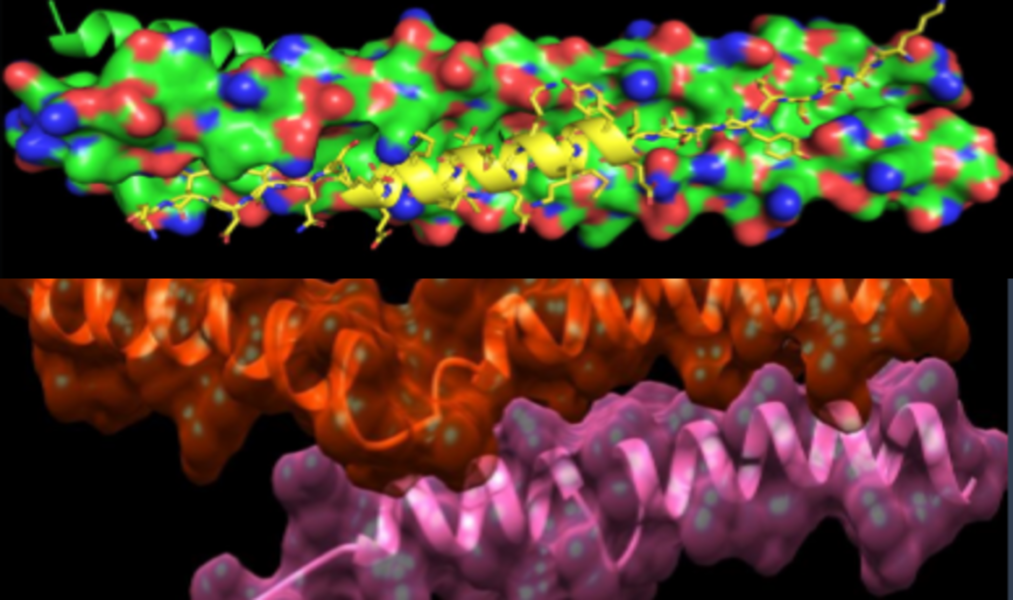
- Use of in silico drug discovery capabilities to identify bioactive peptides.
- 40 peptide analogs were synthesized (4 rounds of iteration), of which
- 15 show S protein binding assessed by GE‘s BIACORE,
- 8 show inhibitory effects on pseudo virus assays,
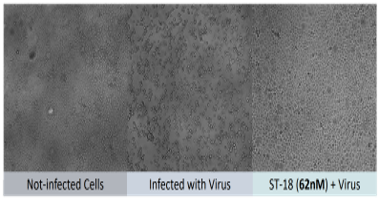
- 3 have inhibitory effects on live Covid-19 / SARS-CoV-2 viruses in a P3 lab
Results and Impacts
- The project time to identify lead molecules (incl. synthesis and in vitro validation on live viruses) was reduced to a few months
- Leveraged know-how and chemoinformatics methods to compensate for incomplete knowledge
- Identification of antiviral peptides with strong S protein binding, potent pseudo virus inhibition and potent live Covid-19 / SARS-CoV-2 inhibition (IC50 = 32 nM)
Janus Kinase (JAK) Inhibitor against Psoriasis
Project Kick-off
Drug design and synthesis
In vitro study
Racemic synthesis and in vitro evaluation
In vivo evaluation
Deliver lead compounds for in vivo experiments
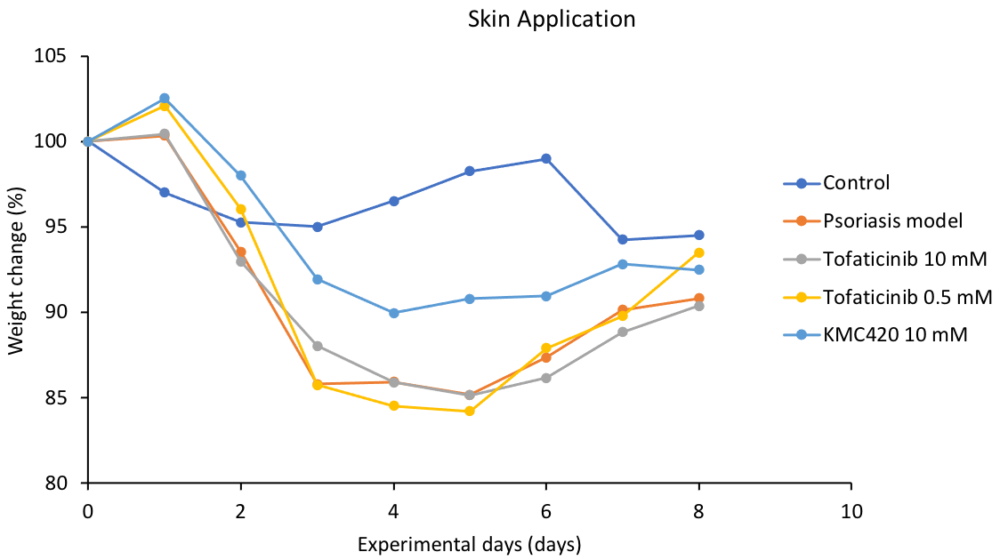
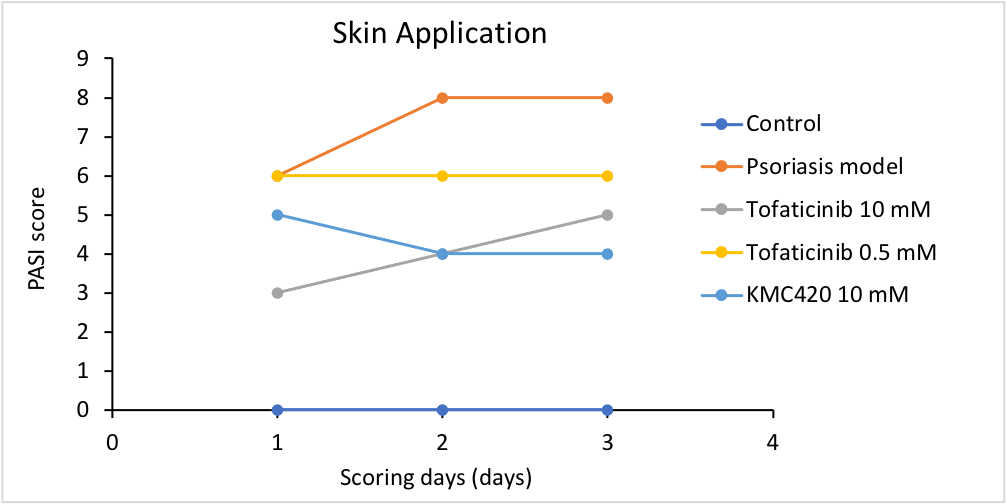
Challenges
Lack of novelty in recent drug development
3D-shaped molecules highly desirable but difficult to synthesize
Lack of selectivity of JAK inhibitor between JAK1, 2 & 3 leading to side effects
Workflow
- Scaffold hopping
- Reference compounds used for drug design, already present in the market or currently in development, and acting as JAK inhibitor: Tofacitinib, Delgocitinib, PF-06651600.
Results and Impacts
- Potentially best-in-class
- Benchmarked to most studied JAK inhibitor: Tofacitinib
- Triquinazine promising in drug design
Antimicrobials peptide drugs (AMPDs) against multi-drug resistant bacteria
Library design & VS
ST-AMPDs SPPS & formulation
In vitro study
Mechanistic studies of membrane integrity
In vivo studies on animal model
Safety and whole blood stability studies
IND
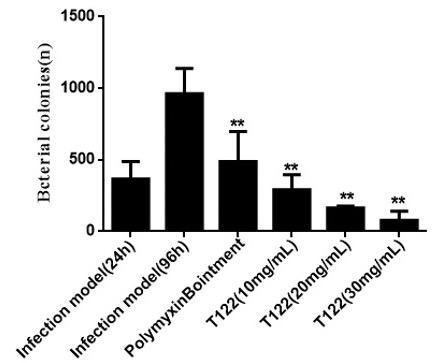
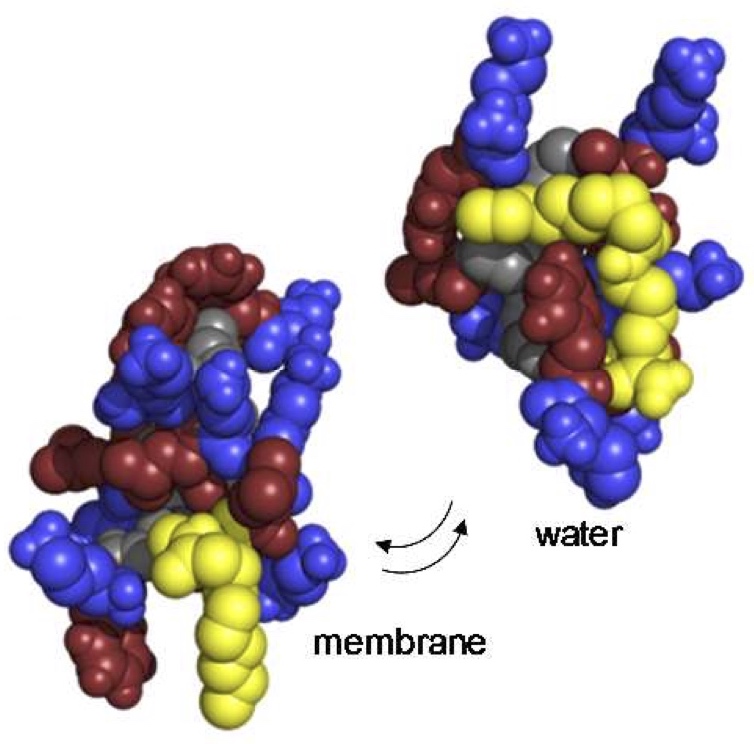
Challenges
- Constant increase in multi-antibiotic resistance in pathogenic bacteria calls for new approaches in antibiotics discovery
- Worldwide shortage of new antibiotics.
- Most existing computer aided design methods are limited to the context of small molecules drugs within the classical Lipinski limits. Hence, the diversity of peptide sequences and topologies are largely ignored. Our technology opens up a whole new area of possibilities in the IP-free chemical space of diverse, non-linear peptides and peptidomimetics.
- Combinatorial chemistry and high-throughput screening are not readily available for either bridged bicyclic or dendrimer peptides
Workflow
- Chemical space guided drug discovery, using the GDBspace computational technology platform
- A virtual library was exhaustively enumerated for generating peptide sequences, and 2DP, a new topological shape and pharmacophore fingerprint for peptides, was calculated for each isomer of each sequence
- From the initial hit identification, further optimization was performed by nearest neighbor search in 2DP space, then clustering and cherrypicking for synthesis and experimental evaluation
- After synthesis, formulation with freeze-dry powder injector, gel or spray was used
- Peptides were tested against a broad-spectrum of clinical strains of MDR-bacteria e.g. P. aeruginosa, E. coli, A. baumannii, S. aureus, K. pneumoniae
- Mechanistic studies of membrane integrity by imaging (Transmission electron microscopy, fluorescence imaging, atomic force microscopy)
Results and Impacts
- First time application of chemical space guided small molecule discovery to the discovery of antimicrobial peptides
- Enabled the logical expansion and exploration of peptide-based drug diversity
- Using only a small number of test compounds, bioactive peptides with unusual topologies were identified
- Patent granted
Peptide inhibitors against anti-macular degeneration
2021.9
2021.10
2021.11
2021.12.7
2021.12.17
2021.12.24
Q2 2022
Kick Off
Drug design & VS
Library design and ultra fast synthesis of 10 compounds
In vitro experiments
Identification of 3 active compounds
Lead compound delivered for in vivo experiment
IND

Challenges
Only biologics present on the market
Workflow
Structure-based drug design
Results and Impacts
- A potentially best-in-class compound was identified
- Benchmarked to first-in-class molecule (AXT-107) currently in Phase II clinical trial
Discovery and optimisation of a selective TRPV6 calcium channel inhibitor
Kick Off
LBVS & clustering
Screening library: 133 Compounds
Hits Optimization
1st micromolar TRPV6 inhibitor
Challenges
TRPV6 is a calcium selective ion channel expressed in many tissues and implicated in several diseases, that has not yet been validated as a therapeutic target due to:
- its poor characterization
- the lack of structure
- the lack of potent and selective inhibitors
Workflow
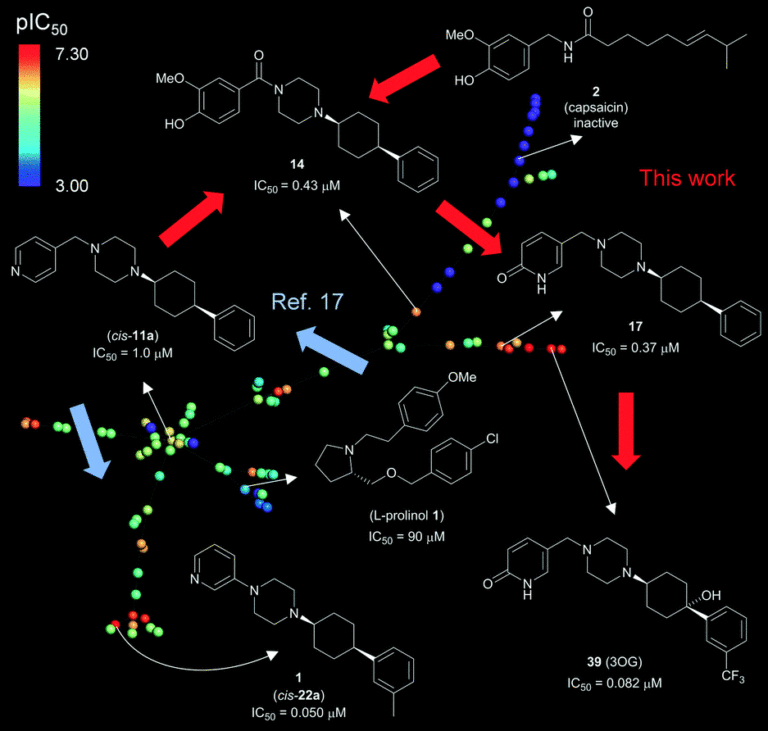
- Fragment-based virtual screening campaign with the 3D shape and pharmacophore similarity search, putting emphasis on scaffold hopping, starting with known weakly active and non-selective TRPV6 inhibitors as seed molecules
- Approx 800,000 purchasable small molecules were scored against seed inhibitors, clustered for diversity, and 133 compounds were purchased for experimental evaluation.
- Further rounds of optimization for achieving high microsomal stability and low off-target activity.
Results and Impacts
- Generated diverse scaffolds and achieved useful hit rates, with as few as a single seed and without target structure information or pre-existing structure-activity relationship data
- Identified the first submicromolar TRPV6 inhibitor, with a seven-fold selectivity against the closely related TRPV5 calcium channel and no activity on store operated calcium channels
- Thanks to its increased potency and selectivity, combined with lack of non-specific toxicity on various cell lines, the new tool compound is now used by multiple labs working on TRPV6
Discovery and optimisation of TRPM4 inhibitors
Kick Off
LBVS & clustering
1st round: 214 Compounds
2nd round: 247 Compounds
SAR Design
1st micromolar TRPM4 inhibitor
Challenges
TRPM4 is a calcium-activated non-selective cation channel expressed in many tissues and implicated in several diseases, that has not yet been validated as a therapeutic target due to:
- its poor characterization
- the lack of structure
- the lack of potent and selective inhibitors
Workflow
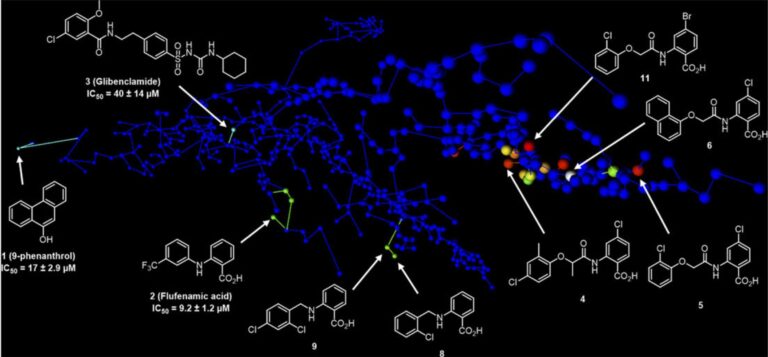
- Ligand-based virtual screening campaign with the 3D shape and pharmacophore similarity search starting with known weakly active and non-selective TRPM4 inhibitors as seed molecules
- Approx 900,000 purchasable small molecules were scored against seed inhibitors, clustered for diversity, and 214 compounds were purchased for experimental evaluation in the first round. In the second round, 247 compounds were selected and purchased for biological testing
- Used MHFP6 chemical space maps to design and interpret SAR
Results and Impacts
- The discovery of a potent and selective TRPM4 inhibitor revealed new opportunities for studying the role of TRPM4 in human diseases
- The SAR studies resulted in two new potent analogs, using MHFP6 chemical space maps for R-group variation tables and for designing, interpreting and communicating the results of this studies
Discovery of a selective Aurora A kinase inhibitor
Kick Off
LBVS & clustering
Library screening: 437 Compounds
2 Hit series identification
Hit optimization
Patent filed
Challenges
Aurora A is a kinases belonging to an evolutionary conserved target family.
- Hurdles in finding novel IP-free compound series in a crowded fields such as kinase inhibitors
- Although many potent inhibitors are known for Aurora A, most also inhibit Aurora B
- Further, the diversity of scaffolds is already high among Aurora A kinase inhibitors (e.g. 174 different scaffolds occurring among 329 inhibitors)
Workflow
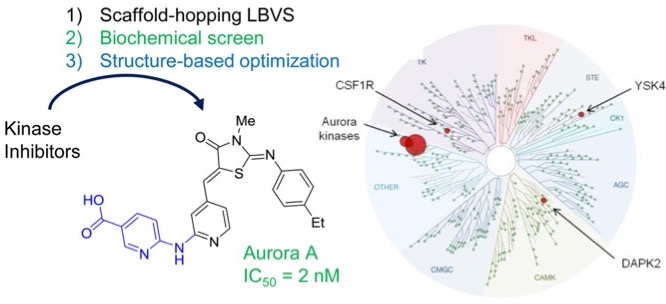
- Used a 3D shape and pharmacophore similarity search of 16 known kinase inhibitors, to select 437 compounds featuring mostly different scaffolds compared to known kinase inhibitors
- Biochemical screening uncovered two inhibitor series with scaffolds unprecedented among kinase inhibitors, one of which was further optimized
Results and Impacts
- Identification of a potent Aurora A inhibitor (IC50 = 2 nM) with very high kinome selectivity.
- The hits identified opens the way to new IP free compound series with high selectivity among evolutionary conserved target family
- The phenotype was rescued by inhibitor-resistant Aurora A mutants. Further, Aurora B specific effects in cells were not induced
- Patent filed
Identification of LPAAT-beta as the target of a nano molar angiogenesis inhibitor from a phenotypic screen
Inhibitor identified in a phenotypic assay
ML based target prediction (PPB2)
1st set of targets (A2aR & VEGFR2) disproved
Target LPAAT-β confirmed
In-cellulo target confirmation
Patent filed
Challenges
- Cytostatic nanomolar inhibitor (an aminotriazine) was identified in a phenotypic co-culture assay
- The compound was initially designed to be a kinase inhibitor but despite its high potency, no kinase activity in a whole kinome profiling assay was observed
Workflow
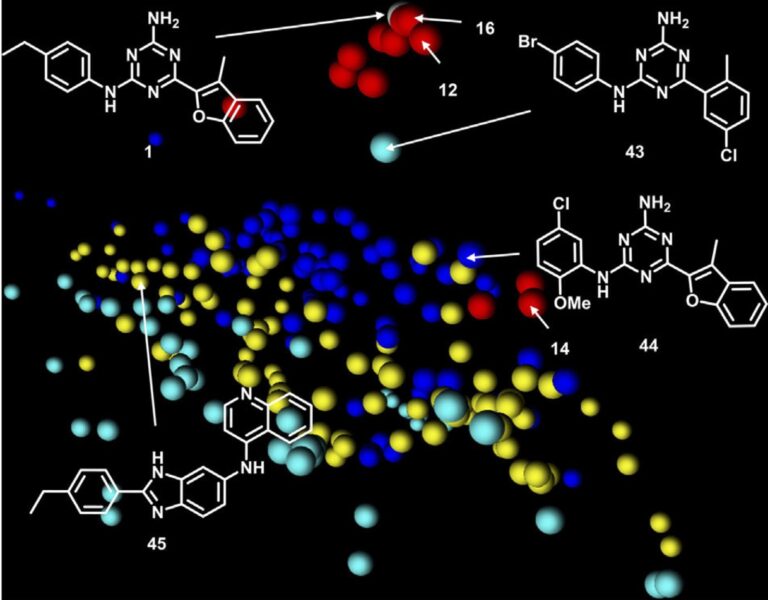
- Machine learning based target prediction (PPB2), which proposes possible targets by similarity to compounds with known ligand-target binding activities
- Target predictions of A2aR and VEGFR-2 were experimentally disproved
- Target assignment of LPAAT-β was confirmed in a biochemical assay and was also consistent with the effect on whole cells in comparison to known LPAAT-β inhibitor
Results and Impacts
- LPAAT-β was identified as a target for the phenotypic screening hits (e.g., this aminotriazine and several analogs), and significantly expanded the rather limited pharmacology of LPAAT-β inhibitors
- Patent filed